The human body is made up of roughly 250 cell types, each defined by particular gene expression patterns.1 An RNA atlas aims to catalog the transcriptome signatures of cells or tissues in their normal state and define potential biomarkers for disease. Concerted research efforts towards this goal include the Human Cell Atlas, the Genotype-Tissue Expression (GTEx) project, Functional ANnoTation Of the Mammalian genome project (FANTOM5), and The Cancer Genome Atlas (TCGA) program.2-7 However, existing consortium data are incomplete. To date, GTEx, FANTOM5, and TCGA have focused on either polyadenylated (polyA) messenger RNA (mRNA) or small RNA and on specific tissues or disease states.
Studies associated with the Human Cell Atlas have used single-cell RNA sequencing (scRNA-Seq) and spatial RNA-Seq methods to map the individual cell types that make up various human tissues and discover new and rare cell types.8-12 Single-cell gene expression offers valuable cell-by-cell resolution in heterogeneous samples, but only a limited view of each cell’s transcriptome.13,14 For a typical scRNA-Seq experiment, 3000 to 6000 transcripts, represented with at least one or two tags, are detected per cell. While this low number of transcripts is sufficient to phenotype cells, it only reveals the most highly expressed mRNAs. Information about the types and amounts of noncoding RNAs (ncRNA) in specific cell types is missing. More comprehensive sequencing is required to access a richer view of the transcriptome.
The importance of a transcriptome encyclopedia
Illumina scientists and collaborators saw the need for a more holistic and inclusive methodology to assemble the ultimate RNA atlas. A deep and thorough transcriptome encyclopedia can be a foundational resource for researchers interpreting scRNA-Seq data and help identify tissues of origin for cancer or other conditions.
Deep sequencing of individual cell types
Through collaborators at the University of Ghent, our combined research team had access to 160 homogenous collections of individual cell types.15 These purified cell populations enabled the benefits of bulk RNA sequencing depth with the focus of single-cell sequencing. The ability to run multiple assays on the same cell-specific RNA samples revealed rich transcriptome complexity and made it easier to resolve background noise from stochastic gene expression. We also sequenced 45 different tissues and 93 cell lines, of which 89 were cancer cell lines derived from 13 different types of cancer. Each sample of the almost 300 distinct tissues and cell types was sequenced very deeply to show virtually every transcript expressed in those cells (Figure 1).
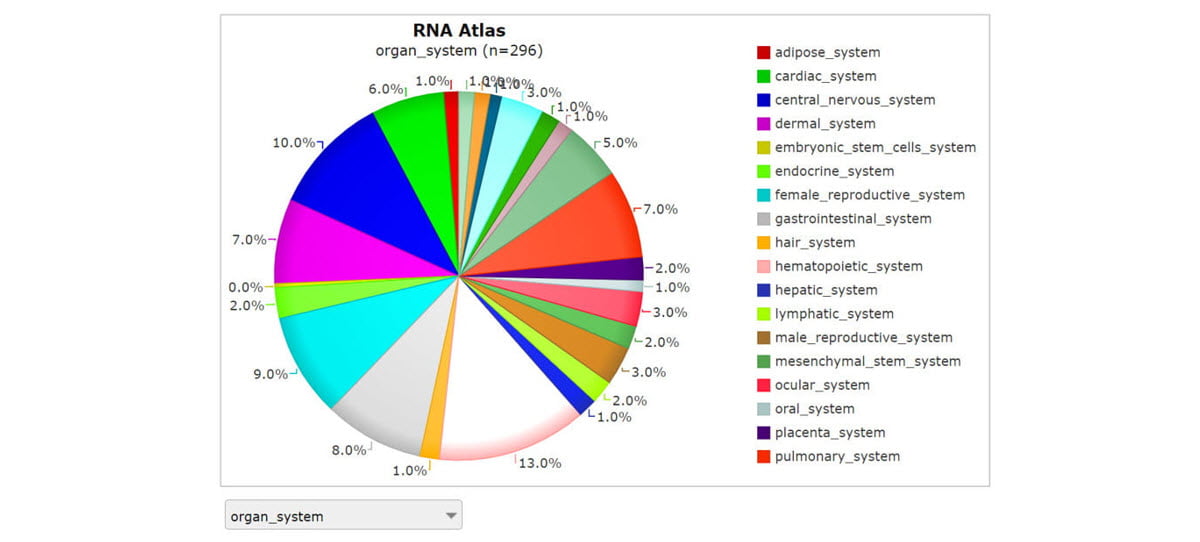
Screenshot from R2: Genomics analysis and visualization platform. The comprehensive RNA atlas study performed strand-specific RNA-Seq for almost 300 distinct tissues and cell types using three complementary library prep approaches: total RNA-Seq, polyA RNA-Seq, and small RNA-Seq.
Comprehensive approach to transcriptome analysis
This study took advantage of three complementary RNA-Seq library preparation approaches (Figure 2) to look at the full transcriptome:
- Stranded total RNA with depletion of ribosomal RNA (rRNA)
- Stranded mRNA with capture of polyA transcripts
- Stranded small RNA to examine micro RNAs (miRNA) and other small ncRNAs
An additional library preparation method, RNA exome enrichment (Figure 2), allowed targeted RNA-Seq from low-input samples, like biofluids.16
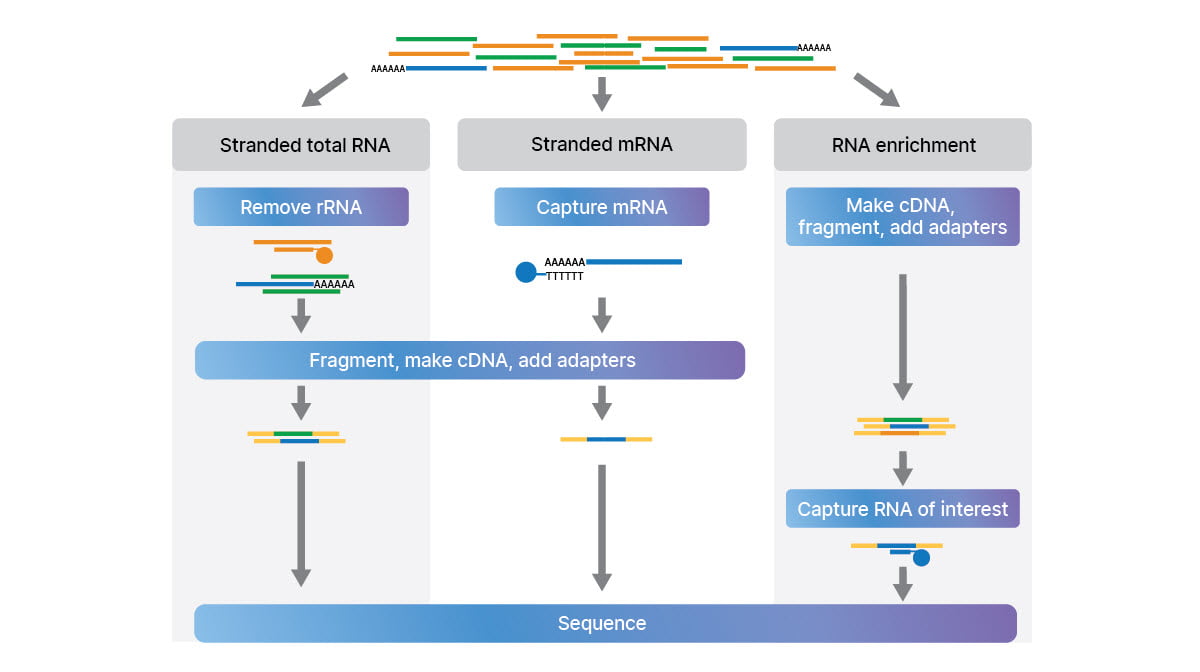
Complementary methods for RNA-Seq library prep: stranded total RNA with rRNA depletion, stranded mRNA with polyA capture, and RNA exome enrichment. Small RNA method not shown.
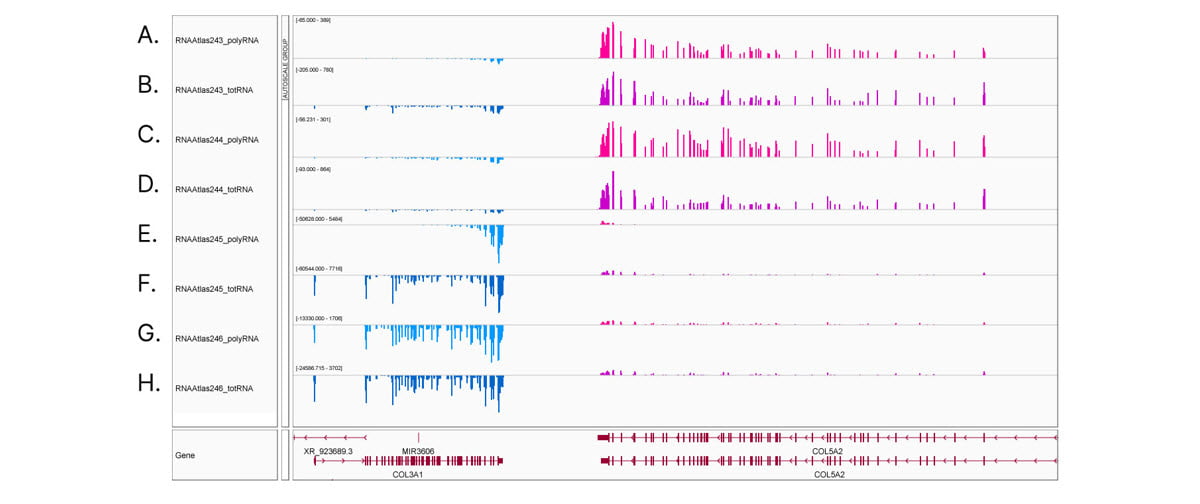
Comprehensive, deep transcriptome analysis demonstrates the importance of stranded RNA-Seq methods. Integrative Genomics Viewer (IGV) screenshot of COL3A1 and COL5A2 gene expression in ocular cell types. Transcripts from the Watson strand are blue, transcripts from the Crick strand are pink. (A,C,E,G) stranded polyA RNA-Seq and (B,D,F,H) stranded total RNA-Seq from (A,B) corneal epithelial cells, (C,D) keratocytes, (E,F) retinal pigment epithelial cells, and (G,H) lens epithelial cells. Read counts shown at auto-scale for each sample track. Corneal epithelial cells and keratocytes show low expression of COL3A1 and high expression of COL5A2. In contrast, retinal pigment epithelial cells and lens epithelial cells show high expression of COL3A1 and low expression of COL5A2. Comparing (E) polyA RNA-Seq to (F) total RNA-Seq in retinal pigment epithelial cells reveals the 3' bias common to polyA RNA-Seq libraries.
Search for novel transcripts
The RNA atlas transcriptomes were annotated against human RNA databases to identify transcripts both known and novel. The comparison data sets included GENCODE, RefSeq, FANTOM5, Comprehensive Human Expressed SequenceS (CHESS), MiTranscriptome, BIGTranscriptome, and several small RNA and ncRNA databases.6,15,17-20 For independent confirmation of novel transcripts, we used transcription start site data from cap analysis of gene expression (CAGE)6 and promoter mapping via chromatin profiles.21 Most of the new transcripts revealed through this RNA atlas study were ncRNAs, including previously predicted intronic and intergenic miRNAs and long intergenic ncRNAs (lincRNA).15 Stranded RNA-Seq was crucial for confirming the validity of single-exon lincRNAs.15
Uncovering a new paradigm in gene regulation
One insight revealed by this comprehensive RNA atlas challenges a traditional dogma about polyadenylation. From this study, more than 75% of lincRNAs are nonpolyadenylated (ie, they were more abundant in the total RNA-Seq than the polyA-selected RNA-Seq library preps). Further, while developing the RNA atlas, we saw thousands of noncoding transcripts that showed a tissue-specific pattern of differential polyadenylation.15 The actual significance of this differential polyadenylation is unknown, but future research can examine its role in gene regulation and potential use as a biomarker for disease states.
Human biofluids atlas
Early detection and origin of disease using easily accessible biofluids would greatly impact treatment options and outcomes. With our collaborators at the University of Ghent, we surveyed RNA in 20 human biofluids ranging from saliva to sweat to breast milk.22 One interesting finding was that seminal fluid and tears were both rich in RNA and generated high-quality sequencing libraries. Human biofluids were also a rich source of circular RNAs (circRNA).22 These RNAs are formed by unique back-splicing events and can function as regulators of gene expression by, for example, binding to miRNAs or regulatory proteins and acting as decoys/sponges. Their circular nature makes them more resistant to nucleases, leading to higher stability in biofluids and increasing their attractiveness as potential biomarkers.23,24
Biofluid profiles largely reflect the tissues that create them. Using the deep insights from the comprehensive RNA atlas, researchers could map RNA from biofluids back to their tissue of origin. For example, seminal fluid was rich in RNA from prostate cells and thus may be a better source for liquid biopsy to screen for prostate cancer than blood. Or RNA from tears could provide information about eye health.
Conclusion
We leveraged our portfolio of RNA library prep solutions to help create two rich resources: a comprehensive human transcriptome atlas and a human biofluids atlas.15,22 These atlases can accelerate scientific discovery by placing a massive and carefully analyzed data set into the hands of other researchers. Subsequent studies can mine these RNA atlas data sets for even greater insights into the expression and regulation of multiple RNA types.
Learn more
Illumina RNA library preparation solutions
Read the papers:
- Lorenzi L, Chiu HS, Avila Cobos F, et al. The RNA Atlas expands the catalog of human non-coding RNAs. Nat Biotechnol. 2021;39(11):1453-1465. doi:10.1038/s41587-021-00936-1
- Hulstaert E, Morlion A, Avila Cobos F, et al. Charting Extracellular Transcriptomes in The Human Biofluid RNA Atlas. Cell Rep. 2020;33(13):108552. doi:10.1016/j.celrep.2020.108552
Explore the RNA atlas data:
Dedicated accessible portal on R2: Genomics analysis and visualization platform
References
- Hatano A, Chiba H, Moesa HA, et al. CELLPEDIA: a repository for human cell information for cell studies and differentiation analyses. Database (Oxford). 2011;2011:bar046. doi:10.1093/database/bar046
- Human Cell Atlas. humancellatlas.org/. Accessed February 15, 2022.
- Lindeboom RGH, Regev A, Teichmann SA. Towards a Human Cell Atlas: Taking Notes from the Past. Trends Genet. 2021;37(7):625-630. doi:10.1016/j.tig.2021.03.007
- Rozenblatt-Rosen O, Shin JW, Rood JE, et al. Building a high-quality Human Cell Atlas. Nat Biotechnol. 2021;39(2):149-153. doi:10.1038/s41587-020-00812-4
- Broad Institute of MIT and Harvard. Genotype-Tissue Expression (GTEx) project. gtexportal.org/home/. Accessed February 16, 2022.
- RIKEN. Functional annotation of the mammalian genome (FANTOM5) project. fantom.gsc.riken.jp/5/. Accessed February 16, 2022.
- National Cancer Institute. The Cancer Genome Atlas (TCGA) program. cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga. Accessed February 16, 2022.
- Wilbrey-Clark A, Roberts K, Teichmann SA. Cell Atlas technologies and insights into tissue architecture. Biochem J. 2020;477(8):1427-1442. doi:10.1042/BCJ20190341
- Deprez M, Zaragosi LE, Truchi M, et al. A Single-Cell Atlas of the Human Healthy Airways. Am J Respir Crit Care Med. 2020;202(12):1636-1645. doi:10.1164/rccm.201911-2199OC
- Luecken MD, Zaragosi LE, Madissoon E, et al. The discovAIR project: a roadmap towards the Human Lung Cell Atlas. Eur Respir J. 2022;2102057. doi:10.1183/13993003.02057-2021
- Haniffa M, Taylor D, Linnarsson S, et al. A roadmap for the Human Developmental Cell Atlas. Nature. 2021;597(7875):196-205. doi:10.1038/s41586-021-03620-1
- Plasschaert LW, Žilionis R, Choo-Wing R, et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature. 2018;560(7718):377-381. doi:10.1038/s41586-018-0394-6
- Mereu E, Lafzi A, Moutinho C, et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nat Biotechnol. 2020;38(6):747-755. doi:10.1038/s41587-020-0469-4
- Chen G, Ning B, Shi T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front Genet. 2019;10:317. doi:10.3389/fgene.2019.00317
- Lorenzi L, Chiu HS, Avila Cobos F, et al. The RNA Atlas expands the catalog of human non-coding RNAs. Nat Biotechnol. 2021;39(11):1453-1465. doi:10.1038/s41587-021-00936-1
- Illumina. Improved detection of circulating transcripts. Published 2021. Accessed February 17, 2022.
- Frankish A, Diekhans M, Ferreira AM, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47(D1):D766-D773. doi:10.1093/nar/gky955
- Frankish A, Diekhans M, Jungreis I, et al. GENCODE 2021. Nucleic Acids Res. 2021;49(D1):D916-D923. doi:10.1093/nar/gkaa1087
- O'Leary NA, Wright MW, Brister JR, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733-D745. doi:10.1093/nar/gkv1189
- Pertea M, Shumate A, Pertea G, et al. CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol. 2018;19(1):208. doi:10.1186/s13059-018-1590-2
- National Institutes of Health. NIH Roadmap Epigenomics Mapping Consortium. The Roadmap Epigenomics Project. roadmapepigenomics.org/. Accessed February 23, 2022.
- Hulstaert E, Morlion A, Avila Cobos F, et al. Charting Extracellular Transcriptomes in The Human Biofluid RNA Atlas. Cell Rep. 2020;33(13):108552. doi:10.1016/j.celrep.2020.108552
- Verduci L, Tarcitano E, Strano S, Yarden Y, Blandino G. CircRNAs: role in human diseases and potential use as biomarkers. Cell Death Dis. 2021;12(5):468. doi:10.1038/s41419-021-03743-3
- Li X, Yang L, Chen LL. The Biogenesis, Functions, and Challenges of Circular RNAs. Mol Cell. 2018;71(3):428-442. doi:10.1016/j.molcel.2018.06.034