This blog is a summary of a published article in Communications Biology (June 2022, PMID: 35794204). We encourage you to read the original article in full. This work was a collaboration between researchers at Illumina Inc., University College London, Baylor College of Medicine, University of Plymouth (UK), University of Dundee (UK), the National Institute of Neurological Disorders and Stroke, and Johns Hopkins University. We thank this international team of scientists for their outstanding contributions to the field!
What is GBA1 and why does it matter?
The GBA1 gene (formerly GBA) encodes a lysosomal enzyme that is responsible for recycling a large molecule present in cell membranes called glucocerebroside. When this gene is inactive, these molecules build up and become toxic to neurons in the brain and other parts of the body.
Inactivation of both copies of GBA1 causes autosomal recessive Gaucher disease, a rare but severe condition characterized by enlarged internal organs and blood cell abnormalities. Full details about Gaucher can be found in GeneReviews.
Individuals with only one active copy of GBA1 are at increased risk of Parkinson disease (PD) and Lewy body dementia (LBD). PD is a movement disorder characterized by tremors, rigidity, slow movements, impaired balance, and sleep disturbance. LBD is characterized by memory loss and impaired cognitive function. Both can be degenerative, meaning that symptoms tend to get worse over time, and lead to significant reduction in quality of life and eventually death. PD is becoming increasingly common as our population ages, with more than 900,000 people estimated to be affected in the United States in 2020. There are many pharmacologic interventions available to people with PD, including levodopa, a dopamine-like molecule that can improve function and slow the course of the disease—but there are no cures.
Along with a variant in the LRRK2 gene, GBA1 variants are the most common identifiable genetic risk factor for PD, which is driving research into drug development and even early clinical trials for gene therapy (see four examples here).
Why is GBA1 a challenging gene to evaluate?
Using standard next-generation sequencing methods, it is possible to have both false positive and false negative results for significant variants in GBA1. This is due to the presence of a highly homologous pseudogene (GBAP1) located near GBA1 on chromosome 1, which confounds standard variant calling approaches and causes genomic instability of this region (Figure 1A). This instability has led to a range of structural variants in human populations arising from either gene conversion or reciprocal recombination (Figure 1B–1D). Some of these converted and recombinant alleles include copy number gains or losses, further complicating genetic analysis. Errors in the human genome reference sequence for GRCh38—three positions in the GBAP1 reference sequence in GRCh38 erroneously contain GBA1 bases—can also cause incorrect results.
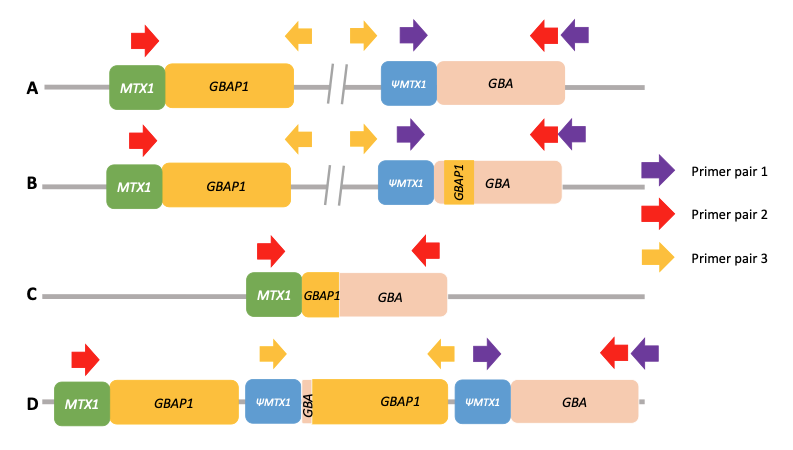
(PMID: 35794204)
A. Wild-type allele. Only PCR with primer pair 1 will produce an amplification. Primer pair 2 will not produce any amplification because the two primers are too far from each other, and primer pair 3 will not produce any amplification because the primers’ orientation does not allow it.
B. Nonreciprocal recombination (gene conversion). Like wild-type alleles, only PCR with primer pair 1 will produce an amplification.
C. Reciprocal crossover between gene and pseudogene resulting in a deletion of a large region of DNA. Only PCR with primer pair 2 will produce an amplification.
D. Reciprocal crossover between gene and pseudogene resulting in the duplication of a large region of DNA. PCR with both primer pair 1 and primer pair 3 will produce an amplification. Note that the normal allele is present and that amplification with primer pair 3 will produce an amplicon independently of the number of copy number gains.
How does DRAGEN resolve these challenges?
The GBA1 targeted caller available in DRAGEN 3.10 and later builds upon the strategies to solve closely related paralogs as described in our previously developed SMN1/2 and CYP2D6 callers.
Using 30× or greater whole-genome sequencing data generated using PCR-free library preparation, this caller calculates the total number of copies of GBA1, GBAP1, and potential GBA1/GBAP1 gene hybrids. The number of reads aligned to the region is normalized and corrected for GC content, and the copy number is called from a Gaussian mixture model. If a copy number alteration is implicated by this analysis, breakpoint detection is performed, taking into account more than 80 differentiating sites containing GBA1 and GBAP1 across the entire region and 10 loci within the most problematic region (exons 9–11 of GBA1).
Ultimately, the GBA1 caller in DRAGEN combines copy number analysis, breakpoint mapping, differentiating site haplotype dosage, and targeted variant detection to genotype all known pathogenic, likely pathogenic, and risk allele variants associated with Gaucher disease, PD, and/or LBD. This includes complex and/or challenging alleles such as E326K, A495P, L483P, N409S, RecNciI, and c.1263del+RecTL (see appendix for details).
How does the GBA1 targeted caller perform?
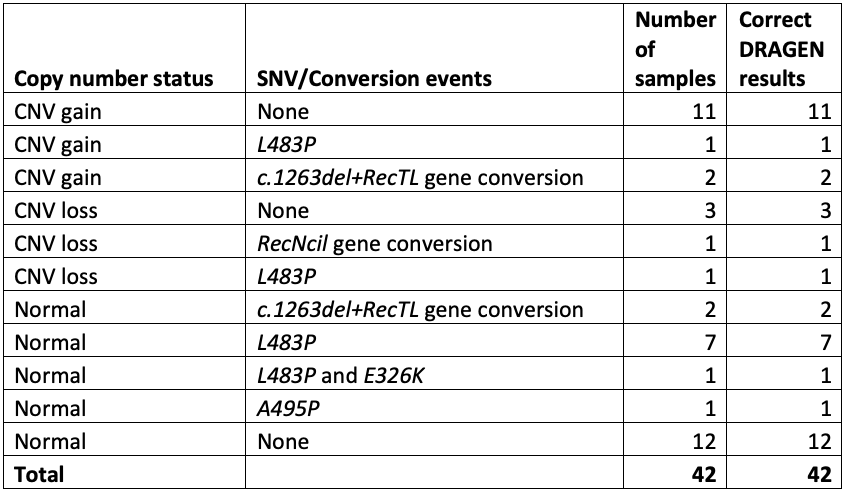
We challenged the DRAGEN GBA1 targeted caller against three increasingly large data sets. For each, we compared the accuracy of DRAGEN against previously established results and/or orthogonal PCR-based testing.
Test 1: 30 cases and 12 controls
The cases included 11 samples with one or more copy number gains, 5 with copy number losses, and 16 with one or more pathogenic small variants. Results from this study were completely concordant for all samples and variants, giving us the confidence to push ahead with further testing (Table 1).
Test 2: Gaucher and Parkinson disease research participants
For this test, we evaluated the performance of the caller on whole-genome sequencing (WGS) data from approximately 400 participants in the RAPSODI trial, roughly split into 50% cases and controls. A subset of these individuals had been tested previously using a PCR-based optical genome mapping approach. Again, the DRAGEN GBA1 targeted caller performed with complete accuracy, correctly identifying 196 positive variants without any false positives (Table 2).
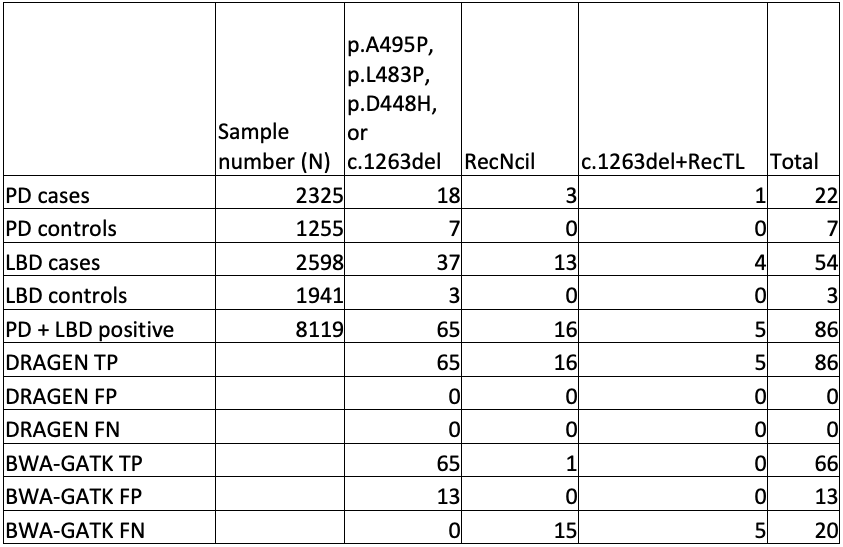
Test 3: 1KGP, AMP-PD, and AMP-LBD data sets
To truly scale up to population-level genomics, we performed GBA1 analysis on publicly available WGS data sets from cases and controls, namely the AMP-PD study and the 1000 Genomes Project (1KGP). These outstanding data sets gave us access to 4923 cases and 5700 control genomes.
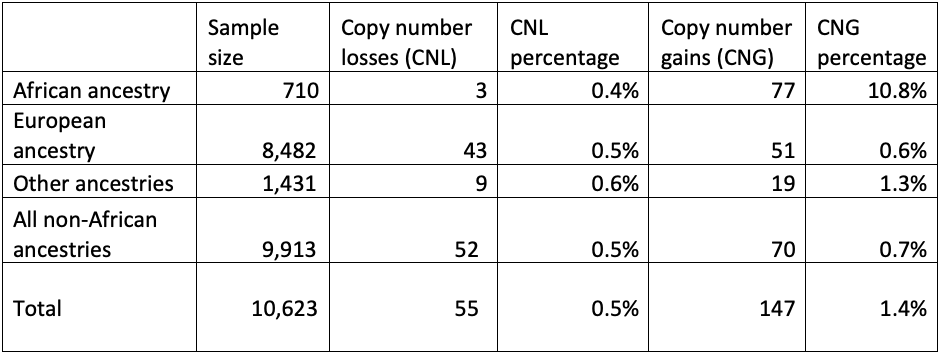
First, we checked the GBA1 copy number status of these genomes, looking for patterns (Table 3). There was no increase of copy number gains or losses in cases versus controls, indicating that these variants are unlikely to contribute to disease risk. However, there was an increase of the proportion of individuals with copy number gains in people who have recent African ancestry compared with those who do not. This highlights the importance of having studies with diverse geographic sample populations, since not knowing this pattern could lead to an incorrect understanding of the molecular pathology at play for GBA1-related conditions. Although these copy number variants are not themselves disease-associated, it is crucial to accurately call the GBA1 copy number status to successfully detect pathogenic small variants and rearrangements.
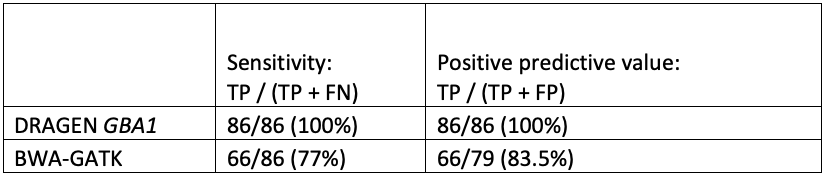
While orthogonal data is not available for all positive results, the improved sensitivity and specificity of GBA1 calling in the difficult exons can be seen in comparing DRAGEN to BWA-GATK (Table 4).
Conclusions
GBA1 is a vitally important gene, and standard methods for its mapping and variant calling are not sufficient. To address this, DRAGEN includes a highly accurate targeted caller for GBA1 capable of cutting through the noise of pseudogene interference and identifying complex gene conversion events in real data. This is yet another example of the DRAGEN team finding ways to maximize the value of PCR-free whole-genome sequencing.
This caller is available now in the latest build of the DRAGEN Bio-IT platform. How would you use this caller to improve your research? What other genes need special attention?
How can I perform GBA1 analysis on my genome data?
There are two ways to take advantage of this targeted caller:
· Bioinformaticians can access the stand-alone version of the caller via GitHub (see README and LICENSE files for details on how to use and cite)
· If you are using DRAGEN v3.10 or later with an on-prem or cloud solution, you can activate the caller as part of the germline DNA sequencing pipeline using “--enable-gba=true”. See online help for further details.
For more information or a DRAGEN trial license for academic use, please contact dragen-info@illumina.com.
Appendix
The following variants are referenced in the text, tables, and/or figure above.
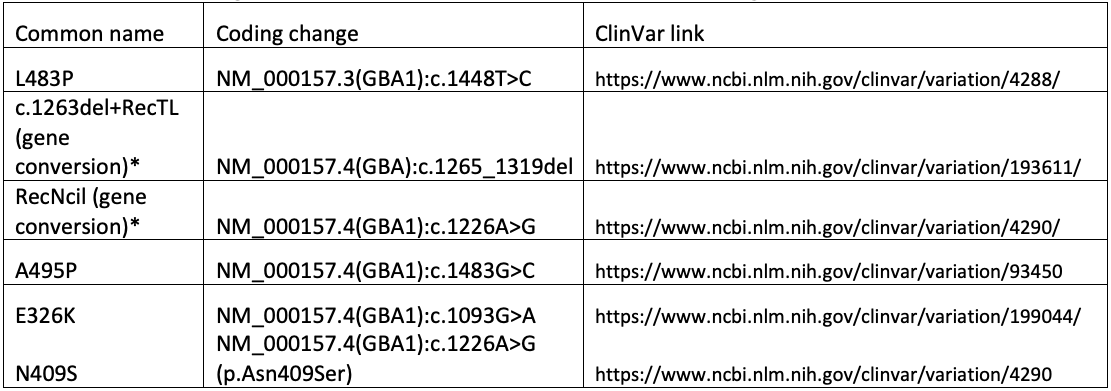
*For gene conversion alleles, a tagging variant is chosen for simplicity. The actual genomic change is more complex than can be readily described using available nomenclature systems.