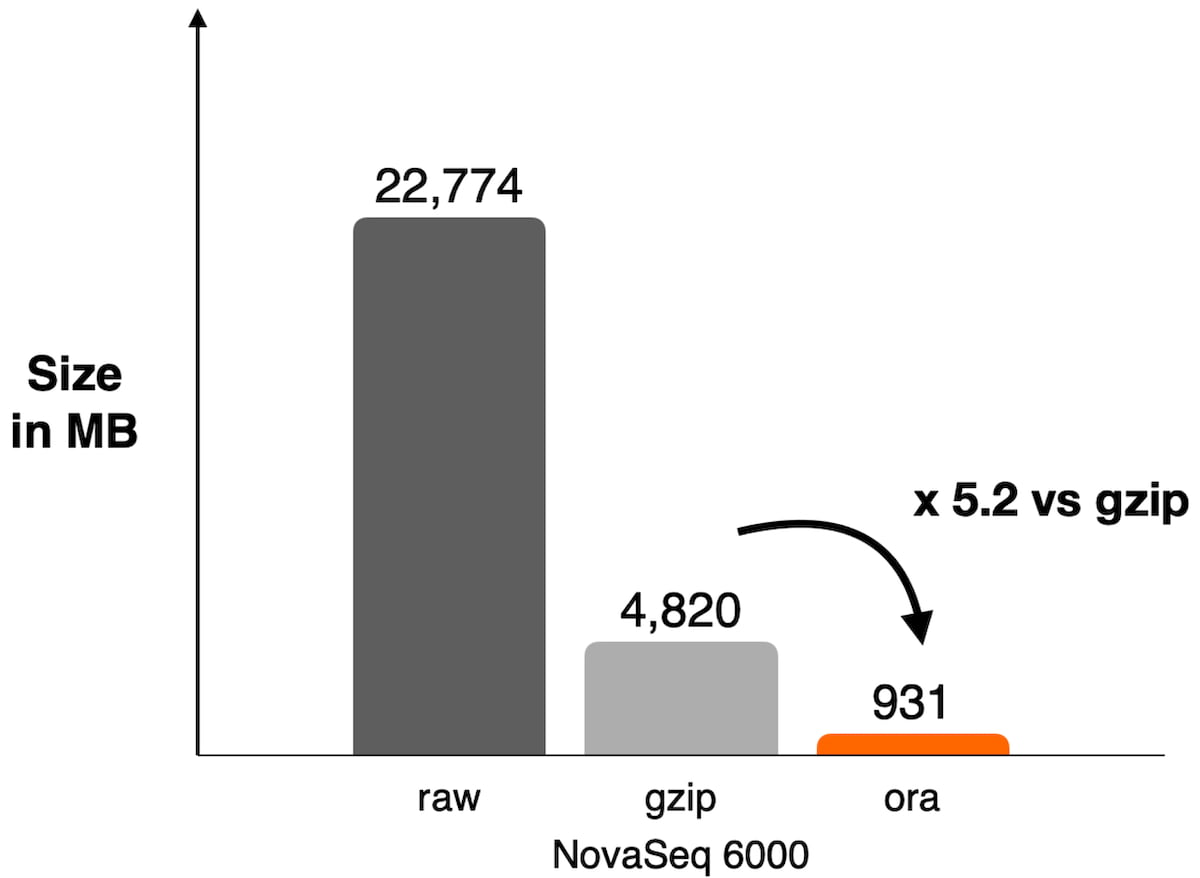
Figure 1: Example of compression ratio reached on NovaSeq 6000 with DRAGEN ORA compression used on WES data SRR8581228
With the volume of data generated with high throughput next-generation sequencing (NGS) increasing exponentially, the FASTQ.ORA format significantly decreases both the storage cost and the energy required to maintain storage servers. To achieve the biggest impact, we need the format to be as widely adopted as possible. In order to favour its adoption, we have released the specification of the format (https://support-docs.illumina.com/SW/ORA_Format_Specification/Content/SW/ORA/ORAFormatSpecification.htm). In this article we will discuss the characteristics of the format that makes it especially well designed for use in an NGS workflow.
Unlike gzip compression, DRAGEN ORA is a compression technology specific to genomic data. The high compression ratio that is achieved is a result from its efficient lossless encoding methodology that we will review further in this article.
Besides efficient encoding, the .ORA format is also designed to allow users to benefit from other advantages:
- Streaming. When downloading an .ORA file, decompression and therefore analysis can start as soon as the first block of reads is downloaded, no need to wait for the whole download to be completed.
- Compressed file concatenation. The format allows for several .ORA files to be written within the same file. This feature simplifies the handling of multiple FASTQ.ORA files.
- Parallel compression / decompression. The design of the format allows for an implementation of the compression/decompression with efficient multithreading in order to accelerate both processes.
- Metadata addition. The format allows for custom metadata to be added.
- Encryption. The format allows for an optional layer of encryption, in order to make the data unreadable without the proper key.
Preamble
The overall objective of this new compression format is to save compute time and money. All design choices are guided by these.
This makes this format different from many compression prototypes produced by academic research, that often aim to explore the boundaries of what is or is not possible e.g., reach as high compression ratio as possible, regardless of the compute cost, or explore the theoretical possibilities of a novel compression method. As examples, Assembltrie1 or lfqc2 aim for high compression ratio but with high compute cost.
1 ORA compression format objectives
There are two “must-have” requirements for the ORA compression format: fully lossless and low-cost integration into NGS workflow.
1.1 Fully lossless
Compression format can be lossy or lossless. For example, for audio, image, or video, it is acceptable to change the original data to allow greater compression. This makes sense since the data modification is not always even perceptible by the user. For genomic data, we require any analysis conducted on compressed data to yield same result as on uncompressed data. The user should have full confidence this requirement is met before launching secondary analysis. The best way to achieve this is to have a fully lossless format, which means that a compression/decompression cycle should preserve data byte-to-byte, so that losslessness can easily be verified with a checksum, such as MD5.
Some existing FASTQ compression formats, such as SPRING3 or ORCOM4, re-order reads since it is an effective method to boost compression. However, some of these formats claim to be lossless on the assumption that initial read order in FASTQ does not carry meaningful information. Even if there is an argument to be made for it to be true, the checksum verification cannot be used anymore to ensure the DNA sequence has been preserved, making it difficult to bring a proof that no information has been lost in the DNA sequence field. Moreover, checksum preservation is in many cases a requirement for quality and regulatory requirements.
For these reasons, we did not choose a read re-ordering method, and made checksum preservation a must-have.
1.2 Low-cost integration into NGS workflow
With saving cost a primary requirement, any compression format that boasts very high compression ratios, but requires significant amounts of memory or compute resources, will not work. Our compression format should not incur unreasonable additional costs and should cause as little disruption as possible in existing analysis workflows.
This implies:
- Low memory usage (in case compression / decompression occurs in parallel to other processes, we should not need a larger system just because of compression)
- Fast enough to not slowdown whole workflow significantly
- Ability to compress / decompress on the fly with no temporary disk files
- Ability to scale to large files
1.3 High compression ratio
We of course want as high compression ratio as possible, but only as long as the first two requirements are met (fully lossless and low-cost integration).
Observations
Academic research in the field showed that compression of FASTQ files can be conducted with reference-free methods (exploiting similarities among reads), or reference-based methods (exploiting similarities between reads and a reference genome).
Reference-free methods can have high compression ratios, especially when combined with read reordering methods, but also tend to have higher execution time or memory usage requirements3,4.
Reference-based methods are easier in principle, such as FQZip5 or LW-FQZip6 – align read to a genome and encode as position plus list of differences – and do not require the use of read re-ordering to achieve high compression ratios. These are possible only when a good quality reference is available. As the vast majority of data produced and stored are and will be human data, choosing a reference-based method for which we have a very good reference available makes sense.
Given our requirements, this led us to choose a reference-based compression method. This seems the most pragmatic choice: it allows for fast execution and high compression ratios, while covering a vast majority of the genomic data produced with the human reference genome used.
2 Description of the FASTQ.ORA compression Format
Many of the design choices were made to reach a good trade-off between speed and compression ratio. The format is also optimized taking into account Illumina sequencing data characteristics: low rate of sequencing errors, no indel errors, short reads.
While the .ORA compression format works with FASTQ data generated with all Illumina platforms, it is most efficient with recent Illumina platforms, e.g. Novaseq 6000 or NextSeq 1000/2000.
2.1 Format layout
An ORA file consists in a list of ORA blocks (Figure 2). Each block contains a chunk of reads and is a self-contained unit that contains all the information needed for its decompression. There is therefore no overall file header; each block has its own header section. This scheme allows for compressed file concatenation, parallel compression/decompression, and streaming capabilities (as soon as a block is transferred, its decompression can start).
Each block holds a chunk of reads, ordered in the same way as the original file. The block starts with a header followed by a list of data sections. The header holds metadata, and the size of each data section. There are separate data sections for DNA sequence, quality scores and read names.
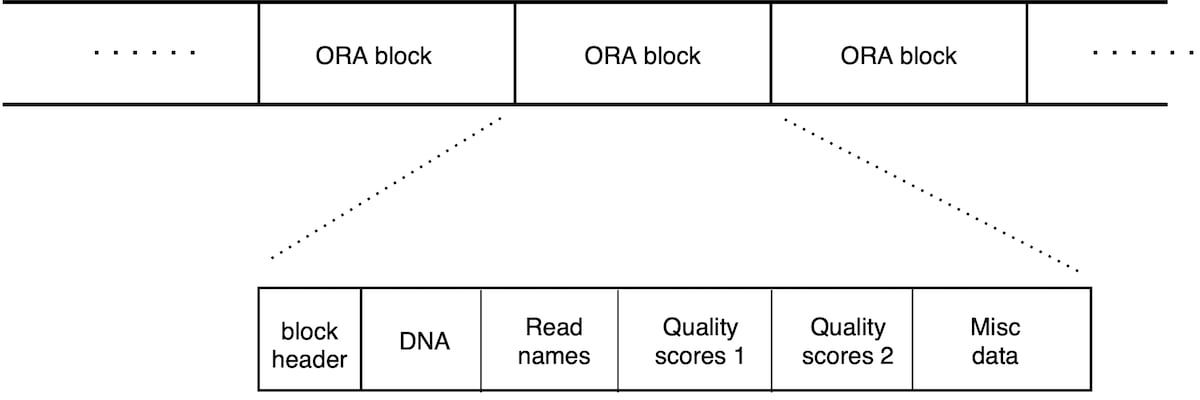
Figure 2: Layout of an .ORA file. List of blocks, each containing 50,000 reads. Each block can be decompressed independently. Each block begins with a header, and then contains different data sections for the DNA sequence, read names and quality scores (that are split in two sections, as explained below).
The layout of an ORA file in Figure 2 shows a sequence of blocks, each containing 50,000 reads. Each block can be decompressed independently. Each block begins with a header, and then contains different data sections for the DNA sequence, read names and quality scores (which are split in two sections, as explained below).
2.2 ORA block
2.2.1 Encoding of DNA sequence
Since we have a reference-based compressor, reads are first mapped to a reference genome, and then encoded as a position in the genome plus a list of differences. The mapping algorithm is not a part of the format specification, it is implementation dependent. To be noted, the choice of read aligner plays an important role in the compression speed and compression ratio. A bit flag in the block header indicates whether reads in the block all have the same size. If not, a list of sizes is stored in a separate data section.
Sequences are encoded as a custom binary format which is then further compressed with an entropy coder, zstd library7.
Reads are encoded with a position on the reference genome (using 32-bit absolute position, or 16 bits offset when possible), and a list of mismatches when necessary. Each mismatch is encoded on one byte, with a 6-bit offset position of the mismatch plus 2-bit alternate nucleotide. When a larger offset is necessary, we add a "fake" mismatch in-between. We allow clipped portions, that are encoded with 2 or 4 bits per nucleotide depending on sequence content. Unmapped reads are encoded raw, also with 2 or 4 bits per nucleotide.
For simplicity and speed, indel sequencing errors are not used. When an indel would have been necessary for optimal alignment, the read will instead be encoded using more mismatches, or with a clipped portion. This does not significantly impact compression ratio since indel sequencing errors are a rare event in Illumina sequencing data (less than 0.03%). Most importantly, omitting indels allows for the use of a very fast mapper.
In the ORA format, we use four different encodings for the reads: perfectly mapped reads that require position only, global alignments that require position + list of mismatches, local alignments that require position + list of mismatches + clipped sequences, and unmapped reads. Some flags indicate for each read the type of encoding used, format of the position (absolute or offset), number of mismatches, and length of clipped portions.
As an example, here is a list of bit sizes for some encodings:
- Perfect alignment, with offset-type position: 4 flag bits + 16 position bits = 20 bits total
- Perfect alignment, with absolute-type position: 4 flag bits + 32 position bits = 36 bits total
- Global alignment, 2 mismatches, absolute position: 12 flag bits + 32 position bits +2*8 bits for mismatches = 60 bits total
- Local alignment, 2 mismatches, 10 nt clipped portion: 92 bits total
- Unmapped 150-nt read: 300 bits
After reads are encoded with this binary format, we then use the zstd library7 to compress further.
After compression (binary encoding + zstd), each nucleotide is encoded in an average of 0.3 to 0.5 bits (compared to 8 bits per nucleotide in raw FASTQ, and about 2 bits / nucleotide in gzipped FASTQ).
2.2.2 Encoding of quality score
For data coming from recent sequencers, quality scores can take only up to 4 different values, with value 0 being used for the quality score of undetermined (‘N’) bases. As a first step, we distinguish between reads that do not contain any 'N’s so have only 3 different quality values, and the rest. We therefore end up with two groups of quality scores, corresponding to the two quality score sections shown Figure 2.
For the group with 3 different quality values, representing the vast majority of reads, we first re-encode quality values with 1.6 bit per quality value (log2(3) ≈ 1.58) and then use a range encoder, with a 4-bit value context model representing the number of highest-value quality scores in the preceding 30-window context.
The sequencer’s binning scheme of quality values affects the relative space quality fields consume in the compressed file as shown in Figure 3. It also has an impact on the compression ratio as shown in Figure 4.
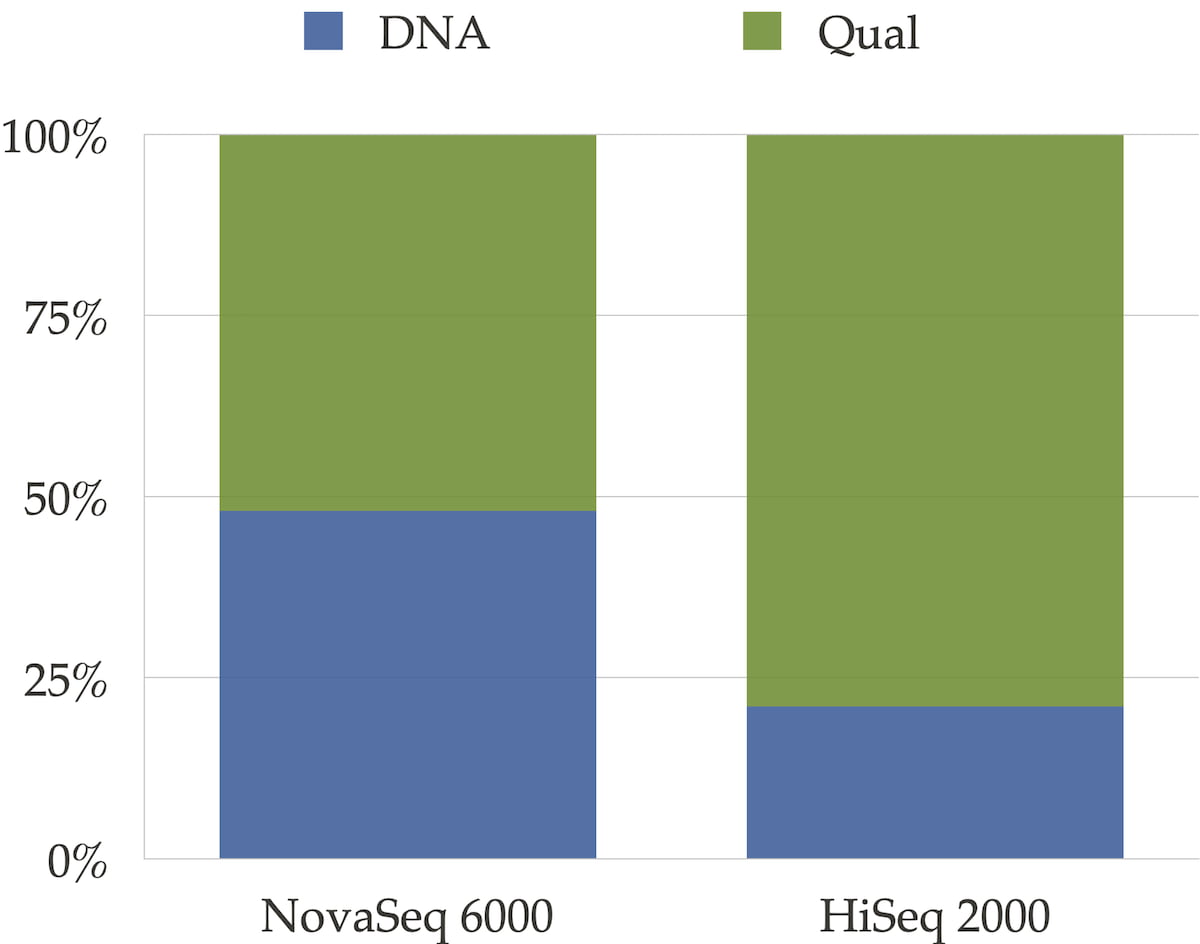
Figure 3: Distribution between DNA and qualities for NovaSeq 6000 and for HiSeqTM 2000 type of data. For the HiseqTM 2000 instrument where quality scores are encoded on 40 different values, qualities take the major part of the file size. For NovaSeq 6000, qualities and DNA take approximately the same space.
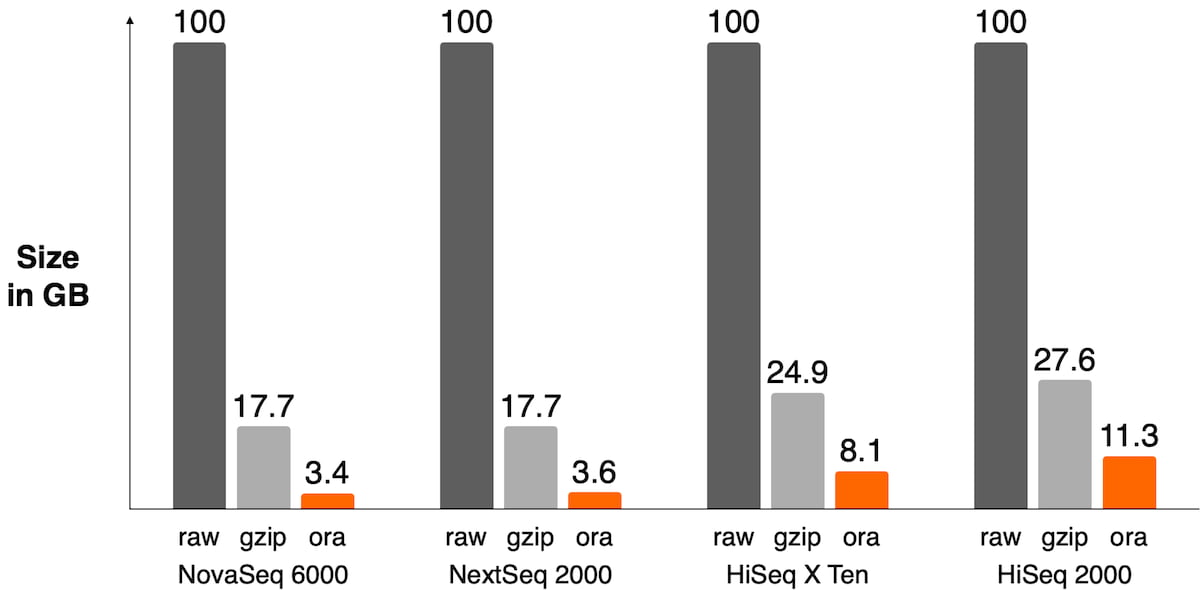
Figure 4: Size of FASTQ.ORA files against raw FASTQ and gzipped files on different sequencers. DRAGEN ORA has the best compression ratio on recent sequencers where quality scores are 2-bit encoded, like on the NovaSeq 6000 or NextSeq 1000/2000 (between 4x and 5x compression ratio vs gzipped file). On older sequencers, the lossless compression of quality scores ends up taking a lot of space in the file and significantly lowers overall compression ratio.
2.2.3 Encoding of read names
Read names are tokenized to separate digits from non-digit fields, and then each token is compressed “column-wise” across all reads of the blocks. Digit fields are encoded as offsets from the previous read, and non-digit with the zstd library.
Several existing FASTQ compression formats already use some variations of this scheme and have shown it to be simple and efficient (e.g. Fqzcomp8).
2.2.4 Integrity checks
Integrity checks are an important part of a compression format. In the ORA format, two checksums are stored within each block of the compressed file:
- Checksum of compressed data: this allows checking for file corruption before the file is decompressed (has the compressed file changed since it was compressed?).
- Checksum of the original uncompressed data: allows verifying that when we decompress, we indeed decompress losslessly.
Integrity checks can be launched manually by the user (i.e. similar to “gzip --test”). In our implementation, the full integrity check is also always performed automatically whenever a file is compressed (when a new compressed block is produced, it is immediately decompressed back and compared to input).
2.2.5 Reference Genome
The default human reference genome currently used in DRAGEN ORA is hg38.
This reference is specific to the compression tool and totally independent from the one that may be used for analysis; it is possible to have completely different reference genome versions for compressing and for secondary analysis mapping.
Moreover, .ORA format allows the use of other reference genomes if needed (although the same reference used at compression will be required at decompression).
3 Resulting performance/ features of the design
DRAGEN ORA compression tool has been designed to meet strict requirements listed above. This design leads to high performance and allows for various convenient features that we describe below.
3.1 Comparison with bzip2 / SPRING
In Table 1 we compare DRAGEN ORA with bzip2 and SPRING3. We do not intend to provide an exhaustive comparison here and chose bzip2 as it is a common alternative to gzip that achieves higher compression ratios, and SPRING as one of the latest FASTQ compression formats published. SPRING was used with default options.
The test is conducted with 16 threads. We compress from a FASTQ.GZ file as input, and decompress back to FASTQ.
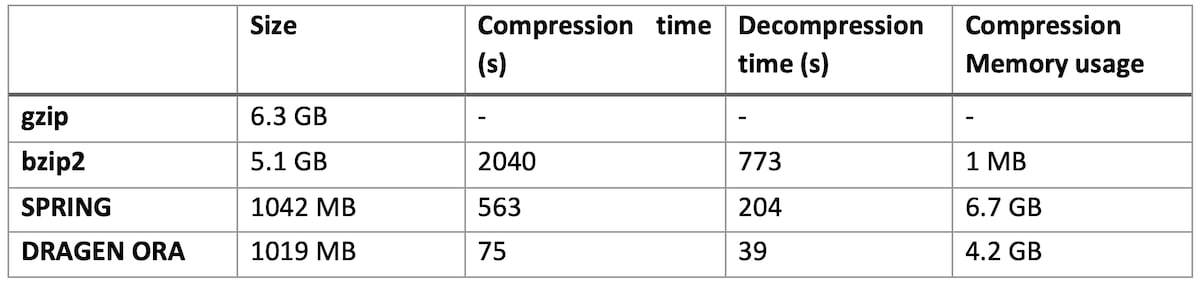
Table 1: DRAGEN ORA performance compared to bzip2, gzip and SPRING on various size files
On file SRR9613620 (human exome sequencing data, with FASTQ.GZ at 6.3 GB)
On file SRR9273189 (human exome sequencing data, with FASTQ.GZ at 14.4 GB)
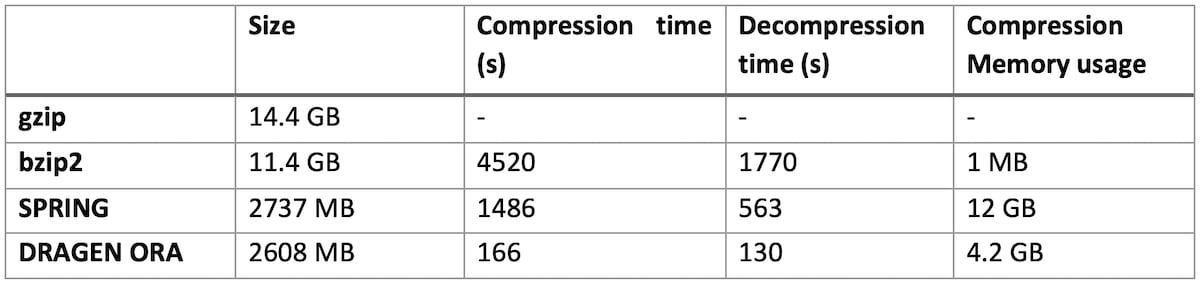
DRAGEN ORA is consistently faster (in the tests above more than 7x faster than SPRING), and still with smaller filer size than SPRING.
Notably, DRAGEN ORA memory usage does not depend on file size, whereas other compressors, e.g., SPRING3, have a memory requirement that grows with the size of input file, making it cumbersome to use on larger files.
3.2 Compression/Decompression speed compared to pigz/gzip
The experiment showed in Table 2 compares DRAGEN ORA compression / decompression speed with pigz, the parallel version of gzip, running with 16 threads on a 7 GB FASTQ.GZ file. In this test, DRAGEN ORA compression is from FASTQ.GZ to FASTQ.ORA, and decompression is from FASTQ.ORA to FASTQ.
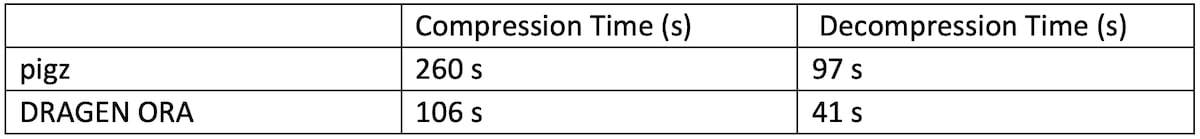
Table 2: DRAGEN ORA speed performance compared with pigz
DRAGEN ORA is faster than pigz both for compression and decompression.
Note that DRAGEN ORA compression (FASTQ.GZ -> FASTQ.ORA) has to unzip the input file before it can compress it into FASTQ.ORA, so unzipping runtime is a lower bound of the DRAGEN ORA compression runtime.
We can see that DRAGEN ORA compression time of 106 seconds is very close to this lower bound (97 s) when running with 16 threads.
3.3 Features enabled by the format specification: streaming, metadata, encryption
The .ORA format is well adapted to streaming usage. When an .ORA file is downloaded, decompression can start as soon as the first block is received. Conversely, when compressing a local file, upload of the compressed file can start as soon as the first block is compressed. DRAGEN v 3.9 features streaming of ORA files from AWS S3 or from Azure Blob Storage.
Additionally, .ORA format has been designed in such a way that encryption can be supported using AES, and ORA implementations are able to write custom metadata in the ORA archive (https://support-docs.illumina.com/SW/ORA_Format_Specification/Content/SW/ORA/ORAFormatSpecification.htm). Note: as of today these features have not been implemented in our current implementation of compression to .ORA format (DRAGEN 3.9 at the time of publication of this article).
We show here that the .ORA format addresses well the requirements of managing high throughput genomic data. The genomics community could greatly benefit from using it. By describing the methodology and sharing the specifications, we hope to encourage its adoption.
Acknowledgements: Thank you to Michael Ruehle and Rami Mehio for their support and for reviewing this article.
For more information or a DRAGEN trial license for academic use, please contact dragen-info@illumina.com.
References
- Ginart, A. A., Hui, J., Zhu, K., Numanagić, I., Courtade, T. A., Sahinalp, S. C., & David, N. T. Optimal compressed representation of high throughput sequence data via light assembly. Nat Commun 9, 566 (2018)
- Nicolae, M., Pathak, S., & Rajasekaran, S. LFQC: a lossless compression algorithm for FASTQ files. Bioinformatics 31, 20, 3276-3281 (2015)
- Chandak, S., Tatwawadi, K., Ochoa, I., Hernaez, M., & Weissman, T. SPRING: a next-generation compressor for FASTQ data. Bioinformatics, 35, 15, 2674-2676 (2019)
- Grabowski, S., Deorowicz, S., & Roguski, Ł. Disk-based compression of data from genome sequencing. Bioinformatics 31, 9 (2015), 1389-1395
- Zhang, Y., Li, L., Xiao, J., Yang, Y., & Zhu, Z. FQZip: lossless reference-based compression of next generation sequencing data in FASTQ format. Proceedings of the 18th Asia Pacific Symposium on Intelligent and Evolutionary Systems (2015) Volume 2 (pp. 127-135). Springer, Cham. DOI: 10.1007/978-3-319-13356-0_11
- Huang, Z. A., Wen, Z., Deng, Q., Chu, Y., Sun, Y., & Zhu, Z. LW-FQZip 2: a parallelized reference-based compression of FASTQ files. BMC bioinformatics, 18, 1, 1-8 (2017)
- https://tools.ietf.org/id/draft-kucherawy-dispatch-zstd-00.html
- Bonfield, J. K., & Mahoney, M. V. Compression of FASTQ and SAM format sequencing data. PloS one, 8, 3, e59190 (2013)